  FIGURA 10-4 FIGURA 10-4
Mecanismos da lesăo tissular induzida pela hiperglicemia. Quatro hipóteses principais sobre como a hiperglicemia causa complicaçőes do diabetes geraram dados abundantes, bem como vários estudos clínicos baseados em inibidores específicos desses mecanismos.
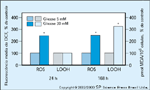 FIGURA 10-5 FIGURA 10-5
A hiperglicemia aumenta as espécies reativas de oxigęnio (ERO) e a peroxidaçăo lipídica (LOOH) intracelulares. Em células endoteliais aórticas, 30 mmol de glicose/L aumentou a formaçăo de ERO (medida como diclorofluoresceína [DCF]) em 250% no período de 24 horas e a peroxidaçăo lipídica resultante (medida como malondialdeído [MDA]) em 330% no período de 168 horas. Portanto, a hiperglicemia aumenta rapidamente a produçăo intracelular de ERO em células afetadas por complicaçőes do diabetes. (Adaptado de Giardino et al. [4].)
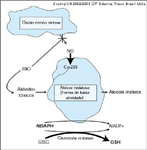 FIGURA 10-6 FIGURA 10-6
Funçăo potencial da aldose redutase em células năo-diabéticas. A enzima aldose redutase converte uma variedade de aldeídos tóxicos (como 2-oxo-aldeídos e aqueles derivados da peroxidaçăo lipídica) em alcoóis inativos. A forma reduzida do fosfato de nicotinamida adenina dinucleotídeo (NADPH) é o cofator tanto nesta reaçăo como na regeneraçăo da glutationa pela glutationa redutase. Espécies reativas de oxigęnio (ERO) parecem reduzir os níveis de óxido nítrico (NO). A atividade da aldose redutase é reversivelmente regulada para baixo ('downregulation') pela modificaçăo pelo óxido nítrico de um resíduo de cisteína no sítio ativo da enzima. GSH — glutationa reduzida; GSSG — glutationa oxidada; NADP — fosfato de nicotinamida adenina dinucleotídeo, forma oxidada. (Adaptado de Chandra et al. [5], Pieper et al. [6] e King e Brownlee [7].)
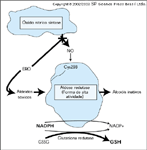 FIGURA 10-7 FIGURA 10-7
Funçăo potencial da aldose redutase em células năo-diabéticas sob estresse oxidativo. Em um ambiente euglicęmico, as espécies reativas de oxigęnio (ERO) aumentam a concentraçăo de aldeídos tóxicos. Ao mesmo tempo, os níveis de óxido nítrico (NO) săo reduzidos, convertendo desta forma a aldose redutase em uma forma de alta atividade. Os níveis da glutationa năo săo afetados. GSH — glutationa reduzida; GSSG — glutationa oxidada; NADP — fosfato de nicotinamida adenina dinucleotídeo, forma oxidada; NADPH — fosfato de nicotinamida adenina dinucleotídeo, forma reduzida. (Adaptado de Chandra et al. [5], Pieper et al. [6] e King e Brownlee [7].)
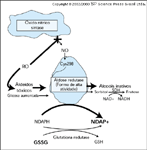 FIGURA 10-8 FIGURA 10-8
Funçăo potencial da aldose redutase em células diabéticas. Em um ambiente hiperglicęmico, as espécies reativas de oxigęnio (ERO) estăo aumentadas, com as mesmas conseqüęncias descritas na Figura 10-7. Além disto, níveis aumentados de glicose intracelular resultam em aumento na conversăo enzimática para poliálcool sorbitol e em reduçőes concomitantes nos níveis da forma reduzida do fosfato de nicotinamida adenina dinucleotídeo (NADPH) e glutationa. Em células nas quais a atividade da aldose redutase é suficiente para depletar a glutationa, o estresse oxidativo induzido pela hiperglicemia seria aumentado. O sorbitol é oxidado a frutose pela enzima sorbitol desidrogenase (SDH). GSH — glutationa reduzida; GSSG — glutationa oxidada; NAD — nicotinamida adenina dinucleotídeo; NADH — nicotinamida adenina dinucleotídeo, forma reduzida; NADP — fosfato de nicotinamida adenina dinucleotídeo. (Adaptado de Chandra et al. [5], Pieper et al. [6] e King e Brownlee [7].)
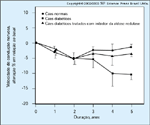 FIGURA 10-9 FIGURA 10-9
Efeito da inibiçăo da aldose redutase sobre diminuiçőes induzidas pelo diabetes na velocidade de conduçăo nervosa. Em căes diabéticos, a conduçăo tornou-se significativamente menor que o normal em 42 meses. A velocidade de conduçăo em căes tratados com inibidores da aldose redutase permaneceu estatisticamente igual ŕ normal durante todo o período de estudo de 5 anos. Em contraste, a inibiçăo da aldose redutase năo teve nenhum efeito sobre o desenvolvimento retinopatia diabética. (Adaptado de Engerman et al. [8].)
 FIGURA 10-10 FIGURA 10-10
Conseqüęncias potenciais da ativaçăo da diacilglicerol-proteína quinase C induzida pela hiperglicemia. A hiperglicemia aumenta o conteúdo de diacilglicerol (DAG), em parte por síntese de novo e possivelmente também por hidrólise da fosfatidilcolina. DAG aumentado ativa a proteína quinase C (PKC), primariamente as formas beta e delta. A PKC ativada aumenta a produçăo de citocinas e matriz extracelular, do inibidor fibrinolítico PAI-1 e do vasoconstritor endotelina-1 (ET-1). A proteína quinase C é também um mediador da atividade do fator de crescimento endotelial vascular (VEGF). Estas alteraçőes contribuiriam para o espessamento da membrana basal, oclusăo vascular, permeabilidade aumentada e ativaçăo da angiogęnese. ANP — peptídio natriurético atrial; eNOS — óxido nítrico sintase endotelial;TGF — fator de transformaçăo do crescimento. (Adaptado de Koya e King [9].)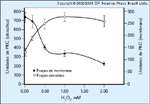 FIGURA 10-11 FIGURA 10-11
As espécies reativas de oxigęnio (ERO) ativam a proteína quinase C (PKC) nas células endoteliais vasculares. Ŕ medida que aumenta o H2O2 produtor de ERO, ele ativa a PKC. O mecanismo parece envolver ativaçăo direta ou indireta da fosfolipase D, que hidrolisa a fosfatidilcolina para produzir diacilglicerol (DAG). As espécies reativas de oxigęnio também poderiam aumentar o DAG através de aumento da síntese de novo resultante da inibiçăo por ERO da enzima gliceraldeído-3-fosfato desidrogenase. (Adaptado de Taher et al. [10] e Schuppe-Koistinen et al. [11].)
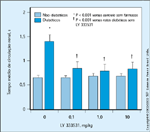 FIGURA 10-12 FIGURA 10-12
Melhora da disfunçăo vascular retiniana induzida pelo diabetes por um inibidor da proteína quinase C beta. Em ratos, o diabetes aumentou o tempo médio de circulaçăo renal de 0,67 segundos para 1,40 segundos. Em pacientes diabéticos, o tratamento com a maior dose do inibidor da proteína quinase C beta, LY 333531, reduziu o tempo para 0,87 segundos. (Adaptado de Ishii et al. [12].)
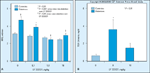 FIGURA 10-13 FIGURA 10-13
Melhora da disfunçăo renal induzida pelo diabetes por um inibidor da proteína quinase C (PKC) beta. Em ratos, o diabetes aumentou o ritmo de filtraçăo glomerular (RFG) de uma média de 3,0 para 4,6 mL/min. Em pacientes diabéticos, o tratamento com a maior dose do inibidor da PKC beta, LY 333531, normalizou a RFG média (A). Similarmente, o diabetes aumentou a taxa de excreçăo de albumina em ratos de uma média de 1,6 para 11,7 mg/d. O tratamento de pacientes diabéticos com a maior dose de LY 333531 reduziu a taxa média de excreçăo de albumina para 4,9 mg/d (B). (Adaptado de Ishii et al. [12].)
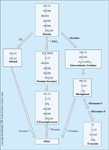 FIGURA 10-14 FIGURA 10-14
Vias potenciais que levam ŕ formaçăo de produtos finais da glicosilaçăo avançada (AGEs – advanced glycation end products). Estes últimos podem originar da auto-oxidaçăo de glicose a glioxal, decomposiçăo do produto Amadori para 3-desoxiglicosona e fragmentaçăo do gliceraldeído-3-fosfato para metilglioxal. Estas dicarbonilas reativas reagem com grupos amino de proteína para formar AGEs. Metilglioxal e glioxal săo destoxificados pelo sistema da glioxalase. Todos os tręs precursores de AGE săo também substratos de outras redutases. (Adaptado de Shinohara et al. [13] e Vander Jagt et al. [14])
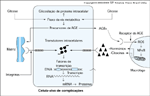 FIGURA 10-15 FIGURA 10-15
A produçăo intracelular de precursores de produto final de glicosilaçăo avançada (AGE – advanced glycation end product) causa lesăo em células-alvo por tręs mecanismos gerais. Primeiro, a glicosilaçăo de proteína intracelular altera a funçăo proteica. Segundo, a matriz extracelular modificada por precursores de AGE tem propriedades funcionais anormais.Terceiro, proteínas plasmáticas modificadas por precursores de AGE ligam-se aos receptores de AGE em células adjacentes, como macrófagos, induzindo assim a produçăo mediada pelo receptor de espécie reativa de oxigęnio (ERO). Esta última, por sua vez, ativa o NFkB e a expressăo de produtos gęnicos patogęnicos, inclusive citocinas e hormônios. mRNA — RNA mensageiro. (Adaptado de Brownlee [15].)
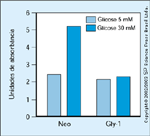 FIGURA 10-16 FIGURA 10-16
A glicosilaçăo proteica intracelular pelo precursor de produto final de glicosilaçăo avançada metilglioxal aumenta a endocitose macro-molecular em células endoteliais. Após uma exposiçăo a 30 mmol de glicose/L, a endocitose macro-molecular por células endoteliais GM7373 que foram estavelmente transfectadas com o gene de resistęncia ŕ neomicina (Neo) foi aumentada em 2,2 vezes. Em contraste, quando um maior acúmulo de metilglioxal foi enviado por uma expressăo excessiva da enzima glioxalase I (Gly-1) nestas células, 30 mmol de glicose/L năo aumentou a endocitose macromolecular. (Adaptado de Shinohara et al. [13].)
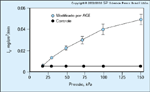 FIGURA 10-17 FIGURA 10-17
A membrana basal glomerular modificada por produtos finais da glicosilaçăo avançada (AGEs – advanced glycation end products) tem uma maior permeabilidade ŕ albumina. A ultrafiltraçăo de albumina (Js) pela membrana basal glomerular modificada por AGE está significativamente aumentada em comparaçăo com a ultrafiltraçăo de albumina pela membrana basal glomerular năo-modificada em uma faixa de diferentes pressőes de filtraçăo. (Adaptado de Cochrane e Robinson [16].)
 FIGURA 10-18 FIGURA 10-18
Proteínas de ligaçăo a produto final de glicosilaçăo avançada (AGE – advanced glycation end product) e supostos receptores. Foram identificadas cinco proteínas de ligaçăo a AGE. 1) RAGE (receptor de AGE) é um novo membro da superfamília das imunoglobulinas; sua ligaçăo gera espécies reativas de oxigęnio e ativa o fator de transcriçăo pleiotrópico NFkB; 2) p60 exibe uma identidade de 95% com o OST-48, um componente do complexo oligossacariltransferase em membranas microssômicas; 3) p90 tem uma homologia significativa de seqüęncia com o 80K-H humano, um substrato da proteína quinase C; 4) galectina-3, uma proteína de ligaçăo a carboidrato, também se liga a AGEs; 5) o receptor scavenger de macrófago de tipo II se liga aos AGEs e medeia sua captaçăo por endocitose. RAGE — receptor de AGE. (Adaptado de Yan et al. [17], Li et al. [18],Vlassara et al. [19] e Araki et al. [20].)
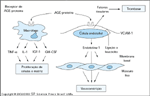 FIGURA 10-19 FIGURA 10-19
Os mecanismos pelos quais a ligaçăo da proteína modificada por produto final de glicosilaçăo avançada (AGE – advanced glycation end product) com os receptores específicos em macrófagos e células endoteliais podem causar alteraçőes patológicas nos vasos sangüíneos diabéticos. Em macrófagos e células mesangiais, a ligaçăo estimula a produçăo de fator-a de necrose tumoral (TNF), interleucina-1 (IL-1), fator-1 de crescimento semelhante ŕ insulina (IGF-1) e fator de estimulaçăo de colônias de granulócitos-macrófagos (GM-CSF) em níveis que aumentam a proliferaçăo de células musculares lisas e aumentam a produçăo de matriz. Nas células endoteliais, a ligaçăo induz alteraçőes procoaguladoras na expressăo gęnica e maior expressăo da molécula-1 de adesăo vascular (VCAM-1) de ligaçăo ao leucócito. (Adaptado de Brownlee [21].)
 FIGURA 10-20 FIGURA 10-20
Melhora das anormalidades em tecidos-alvo diabéticos por um inibidor de produto final de glicosilaçăo avançada. Os efeitos deste inibidor (aminoguanidina) sobre anormalidades diabéticas foram investigados na retina, rim, nervo e artéria. Em animais experimentais, o desenvolvimento de todas as anormalidades patognomônicas examinadas foi inibido em 85% a 90%. Em um grande estudo randomizado, duplo-cego, controlado por placebo e multicęntrico de pacientes diabéticos tipo 1 com nefropatia patente, o tratamento com aminoguanidina reduziu a proteína urinária total e desacelerou a progressăo da nefropatia. Além disto, a aminoguanidina reduziu a progressăo da retinopatia diabética (Bolton et al., dados năo-publicados). (Adaptado de Brownlee [15].)
voltar |
















;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)