  FIGURA 14-6 FIGURA 14-6
Síndrome de Cushing. A síndrome de Cushing é uma causa endócrina comum de intolerância ŕ glicose e diabetes secundários. A homeostase anormal da glicose pode resultar da administraçăo exógena diária ou em dias alternados de glicocorticóides ou como conseqüęncia de excesso endógeno crônico de glicocorticóides causado por hipersecreçăo hipofisária do hormônio adrenocorticotrófico (ACTH), produçăo paraneoplásica de ACTH por células tumorais, ou hiperfunçăo autônoma de córtex adrenal. Esta última afetou esta paciente, fotografada antes (A) e depois (B) da apresentaçăo com síndrome de Cushing fenotípica. Foi constatado que a paciente tinha um adenoma de córtex adrenal.
Embora hiperglicemia de jejum ocorra em aproximadamente 5% dos pacientes com síndrome de Cushing, a resistęncia ŕ insulina com hiperinsulinemia basal ou estimulada ocorre em até 90% dos pacientes. Os pacientes com síndrome de Cushing podem se apresentar em um estado hiperosmolar, năo-cetótico [1]. Esta apresentaçăo é extremamente rara em pacientes com acromegalia, que, por outro lado, exibem um espectro de perfis anormais de glicose e insulina semelhantes ŕqueles de pacientes com a síndrome de Cushing. (Cortesia de Jaishree Jagirdar, Escola de Medicina da Universidade de Nova York, Nova York.) 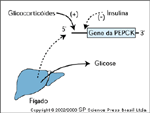 FIGURA 14-7 FIGURA 14-7
O excesso de glicocorticóide promove produçăo hepática de glicose. Várias enzimas-chaves que controlam a produçăo e a utilizaçăo de combustíveis metabólicos săo diretamente reguladas por glicocorticóides no nível da transcriçăo gęnica. A fosfoenolpiruvato carboxiquinase (PEPCK), uma enzima crucial na neoglicogęnese, é regulada positivamente por glicocorticóides [10]. De fato, camundongos transgęnicos que superexpressam PEPCK exibem tolerância ŕ glicose diminuída [11]. Além disto, a exposiçăo de ratas prenhas ŕ glicocorticóide em excesso, no final da gestaçăo, aumenta permanentemente a expressăo hepática de PEPCK e receptor de glicocorticóide e causa intolerância ŕ glicose nos descendentes adultos [12]. Sob condiçőes fisiológicas normais, contudo, a insulina regula PEPCK de forma mais potente e dominante, de uma maneira negativa [13]. Portanto, insulina farta previne a neoglicogęnese aumentada que seria causada por excesso de glicocorticóide. Na presença de deficięncia de insulina ou de comprometimento da açăo da insulina, porém, os efeitos estimulantes dos glicocorticóides sobre a produçăo de glicose tornam-se aparentes. Já que os pacientes com Síndrome de Cushing quase sempre apresentam resistęncia ŕ insulina, o palco fica preparado para a neoglicogęnese amplificada por glicocorticóide, que contribui para a intolerância ŕ glicose.
 FIGURA 14-8 FIGURA 14-8
Glicocorticóide em excesso diminui a utilizaçăo periférica de glicose. Os glicocorticóides induzem a captaçăo de glicose estimulada por resistęncia ŕ insulina em adipócitos de rato [14]. Este efeito pode ser, pelo menos em parte, mediado pela inibiçăo direta por glicocorticóide da translocaçăo induzida pela insulina da proteína quinase C, do citosol para a membrana plasmática. Os glicocorticóides também inibem a ativaçăo do transporte de glicose em músculo esquelético de rato pela insulina, fator-1 de crescimento semelhante ŕ insulina e hipóxia [15]. No músculo soleus de rato, este efeito está associado ŕ preservaçăo do conteúdo total dos transportadores de glicose GLUT4, mas também ŕ menor translocaçăo de unidades do transportador GLUT4 até a membrana plasmática [16]. Além destes efeitos sobre o tráfego subcelular de GLUT4, os glicocorticóides afetam as etapas iniciais da sinalizaçăo do receptor de insulina no músculo esquelético e no fígado [17]. Conseqüentemente, a captaçăo e a utilizaçăo de glicose, tanto basais quanto estimuladas pela insulina, estăo sujeitas ŕ modulaçăo pelos glicocorticóides em excesso.
 FIGURA 14-9 FIGURA 14-9
Os glicocorticóides inibem a secreçăo de insulina das células b pancreáticas. A resistęncia ŕ insulina é reconhecida há muito tempo como uma conseqüęncia do excesso de glicocorticóide. Os efeitos dos glicocorticóides sobre a secreçăo de insulina in vivo e in vitro, contudo, só foram descritos recentemente. Camundongos transgęnicos superexpressando o receptor de glicocorticóide (GR – glucocorticoid receptor) sob o controle do promotor da insulina aumentaram a sensibilidade ao glicocorticóide que está restrita ŕs células b pancreáticas [18]. Estes animais tęm níveis normais de glicose sangüínea em jejum e pós-absortiva, mas também tęm uma resposta de insulina acentuadamente reduzida e tolerância ŕ glicose diminuída durante uma carga de glicose intravenosa. Esta evidęncia in vivo que sugere um efeito diabetogęnico dos glicocorticóides sobre as células b pancreáticas é corroborada por evidęncias in vitro de degradaçăo pós-traduçăo induzida por dexametasona dos transportadores de glicose GLUT2 de células b [19] e de inibiçăo induzida pela dexametasona da liberaçăo exocítica de insulina das ilhotas de roedores em cultura [20]. Uma utilizaçăo diminuída de glicose na síndrome de Cushing pode, portanto, ser um resultado composto de secreçăo deficiente de insulina e comprometimento da açăo da insulina.
voltar |
















;)
;)
;)
;)