
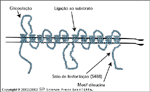
 FIGURA 2-10 FIGURA 2-10
Transportadores de glicose. O transporte de glicose é mediado por uma família de carregadores facilitadores de glicose, ou transportadores de glicose. Estas proteínas tęm uma estrutura típica, que inclui um aminoterminal citoplasmático seguido de 12 domínios que se estendem pela membrana e uma cauda citoplasmática. É mostrada a estrutura proposta do transportador-4 de glicose responsivo ŕ glicose (GLUT4). A primeira alça exofacial contém sítios de glicosilaçăo, ao passo que o domínio citoplasmático carboxiterminal contém um sítio putativo de fosforilaçăo (serina 488) e um motif dileucina que se acredita que desempenha um papel na rápida endocitose do GLUT4. É também mostrado o sítio proposto de ligaçăo ao substrato [48]. Existem evidęncias de oito membros diferentes da família de genes de transportadores de glicose. GLUT1 é expresso em todos os tecidos e linhagens celulares e responde pela maior parte da captaçăo basal (insulino independente) de glicose. GLUT2 é expresso principalmente no fígado e células b pancreáticas; por causa de sua afinidade relativamente baixa e sua alta capacidade pela glicose, ele serve para proporcionar um fluxo constante de glicose para dentro desses órgăos em concentraçőes plasmáticas fisiológicas de glicose (5 mM). Na célula b, a captaçăo de glicose através de GLUT2 é a primeira etapa na detecçăo de níveis circulantes de glicose. GLUT3 é expresso ubiquamente e possui uma afinidade relativamente elevada pela glicose. Curiosamente, GLUT3 é abundante no sistema nervoso central, onde as concentraçőes de glicose săo menores que na corrente sangüínea. A presença de um transportador de glicose de alta afinidade oferece um mecanismo para uma captaçăo eficiente de glicose pelos neurônios. GLUT4 é o transportador de glicose responsivo ŕ insulina prototípico. GLUT4 é encontrado em vesículas intracelulares em tecidos responsivos ŕ insulina, que incluem músculo esquelético, células adiposas e coraçăo. Existem atualmente evidęncias da presença de mais um transportador de glicose responsivo ŕ insulina, GLUT8. GLUT5 é um transportador de frutose expresso na membrana epitelial intestinal e rim. GLUT6 é um pseudogene năo-funcional. GLUT7 é um transportador microssômico de glicose que faz parte do complexo glicose-6- fosfatase que se acredita que desempenhe um papel na liberaçăo de glicose do retículo endoplasmático [48].
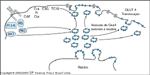 FIGURA 2-11 FIGURA 2-11
Açăo da insulina no transporte de glicose. Nos tecidos que săo responsivos ŕ insulina para captaçăo de glicose, GLUT4 está presente em condiçőes basais nas vesículas intracelulares. Vesículas contendo GLUT4 săo liberadas de compartimento de depósito intracelular e atingem a superfície celular durante a estimulaçăo insulínica. O mecanismo de redistribuiçăo de transportadores de glicose induzida pela insulina é conhecido como translocaçăo. Năo se sabe se o efeito da insulina de estimular a captaçăo de glicose no músculo e na gordura pode ser explicado integralmente pela hipótese da translocaçăo. Outros fatores, como um aumento na atividade de transportadores de glicose, poderiam desempenhar um papel. Existem evidęncias substanciais que sugerem que a atividade da fosfatidilinositol 3-quinase (PI 3-K) medeia o efeito da insulina sobre o transporte de glicose. Além da atividade da PI 3-K, outros sinais parecem ser necessários para a captaçăo de glicose estimulada pela insulina, inclusive uma segunda via envolvendo a fosforilaçăo em tirosina do proto-oncogene de Cbl [49]. Na maioria das células responsivas ŕ insulina, a Cbl está associada ŕ proteína adaptadora CAP, que se liga a seqüęncias ricas em prolina na Cbl através de seu domínio SH3 caboxiterminal. Após fosforilaçăo, o complexo Cbl–CAP se associa ŕ flotilina nos ‘rafts’ (‘jangadas’) lipídicos de membrana plasmática onde a fosfo-Cbl recruta o complexo CrkII-C3G. O CrkII forma um complexo constitutivo com a C3G, uma proteína de troca de guanil nucleotídeo que catalisa a troca de GTP por GDP na proteína-G TC10. A TC10 ativada pode contribuir para a estabilizaçăo da actina cortical e fornecer ŕ proteína GLUT4 um segundo sinal que funciona em paralelo com a ativaçăo da via da PI 3-K.
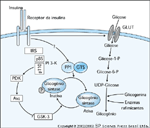 FIGURA 2-12 FIGURA 2-12
Estimulaçăo da síntese de glicogęnio pela insulina. A insulina desempenha um papel essencial na estimulaçăo da síntese de glicogęnio em muitos tecidos. Este processo envolve a regulaçăo de várias atividades enzimáticas. A atividade da glicogęnio sintase (GS) é regulada por fosforilaçăo. Esta enzima crucial é inativa quando fosforilada [50]. A glicogęnio sintase quinase-3 (GSK-3) é uma enzima que catalisa a fosforilaçăo e a inativaçăo da glicogęnio sintase. A própria GSK-3 é inativada por fosforilaçăo. A insulina estimula a fosforilaçăo da GSK-3 através da ativaçăo da Akt quinase (produto do protoonco-gene de akt). A Akt é ativada em resposta ŕ fosfatidilinositol 3-quinase (PI 3-K) através das quinases-1 e 2 dependentes de PI (PDK1 e PDK2). A Akt pode fosforilar e inativar a GSK-3, reduzindo portanto a taxa líquida de fosforilaçăo da GS [51]. Adicionalmente, foi demonstrado que a insulina aumenta a atividade da forma ligada ao glicogęnio da serina/treonina proteína fosfatase-1 (PP1) que desfoforila e ativa a GS [52]. O mecanismo exato da estimulaçăo de PP1 pela insulina năo foi ainda elucidado [53]. A síntese de glicogęnio exige a montagem de um complexo de proteínas, inclusive a glicogenina, para fornecer um arcabouço molecular, enzimas ramificantes e uma variedade de subunidades com alvo dirigido ao glicogęnio (GTS – glycogen targeting subunits) que regulam a localizaçăo e atividade da PP1 [54]. Estudos atualmente em andamento investigam até que ponto a insulina regula a localizaçăo e/ou funçăo da GTS. Foi também demonstrado que o transporte de glicose estimulado pela insulina contribui para a regulaçăo da GS [55].
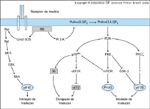 FIGURA 2-13 FIGURA 2-13
Regulaçăo da síntese proteica pela insulina. A insulina estimula tanto a iniciaçăo quanto a elongaçăo, alterando a atividade intrínseca ou as propriedades de ligaçăo de certos fatores-chave. Isto ocorre via fosforilaçăo ou seqüestro de fatores repressivos em um complexo inativo. Os componentes da maquinaria de traduçăo que săo alvos da regulaçăo insulínica incluem eIF2B, eIF4E, eIF4G, S6, eEF1 e eEF2 [56]. O glicogęnio sintase quinase-3 (GSK-3) fosforila eIF2B em Ser540, mantendo-a inativa. A insulina estimula a inibiçăo da GSK-3 que, por sua vez, leva ŕ ativaçăo de eIF2B [48,50]. A insulina também estimula a atividade da eIF2B através da ativaçăo dependente de PI 3-quinase da proteína quinase C atípica, PKCz [57]. Esta ativaçăo da eIF2B leva a um aumento global na iniciaçăo da traduçăo [58]. A proteína de ligaçăo ao cap do mRNA (mRNA cap-Ras binding protein), eIF4E, é inativa quando ligada ŕ PHAS-I. A insulina ativa a eIF4E estimulando a fosfo-Grb2- rilaçăo mediada por mTOR da PHAS-I, resultando numa dissociaçăo deste complexo [59]. A Mnk é ativada pela insulina através da cascata do Ras/ERK [60]. A Mnk aumenta a afinidade da eIF4E pelos caps via fosforilaçăo em Ser209 [61]. A fosforilaçăo e subseqüente afinidade da eIF4G, outro membro do complexo ativo de ligaçăo ao cap (active cap-ERK binding complex) pela eIF4E, é também estimulada pela insulina [62,63]. A formaçăo do complexo ativo de ligaçăo ao cap leva a um aumento da iniciaçăo da traduçăo. A insulina também estimula a elongaçăo pela fosforilaçăo de eEF1 e S6, a última ocorrendo tanto pela S6 quinase multipotencial quando pela p70s6K. eEF2, um importante fator no processo de elongaçăo, é inativa quando fosforilada [64]. A insulina estimula a desfosforilaçăo da eEF2 via uma rota sensível ŕ rapamicina, envolvendo possivelmente a fosforilaçăo e a inativaçăo da eEF2 quinase pela p70s6K [65]. Outros fatores de crescimento agindo através de tirosina quinases do receptor produzem alteraçőes semelhantes na síntese proteica, eg, fator de crescimento derivado de plaqueta, fator de crescimento neural e fator de crescimento epidérmico.
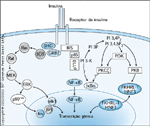 FIGURA 2-14 FIGURA 2-14
Regulaçăo da expressăo gęnica pela insulina. A insulina efetiva a expressăo gęnica-alvo em diversas células e órgăos. Foi demonstrado que as vias de sinalizaçăo tanto da fosfatidilinositol 3-quinase (PI 3-K) quanto da Ras/quinase de regulaçăo extracecular (ERK – extracellular-regulated kinase) medeiam a açăo da insulina sobre a transcriçăo gęnica em diferentes tecidos. A via Ras/ERK ativa a p90rsk, pela qual a ERK e a p90rsk translocam-se individualmente ao núcleo e fosforilam os fatores de transcriçăo, inclusive elk1 e fos. Estes eventos de fosforilaçăo aumentam a atividade transcricional mediada por tais fatores. A via PI 3-K ativa a proteína quinase B (PKB) através da proteína quinase dependente de PI (PDK). A PKB ativa fosforila membros da família do enhancer transcricional FKHR, que inclui FKHR-L1 e HNF3, os quais, sob condiçőes basais, estăo localizados no núcleo. A fosforilaçăo destes fatores FKHR leva ŕ sua exclusăo nuclear e retençăo citoplasmática, inibindo efetivamente a transcriçăo gęnica específica [66]. A insulina também efetiva a dissociaçăo do complexo inibitório de IkB da NF-kB. NF-kB estará entăo livre para entrar no núcleo e ativar a transcriçăo gęnica-alvo. Foi demonstrado que a estimulaçăo da NF-kB pela insulina é sensível a inibidores da PI 3-K, implicando o envolvimento da via da PI 3-K [67]. A ativaçăo da proteína quinase C atípica, como a PKCz, também pode contribuir para a ativaçăo da NF-kB. A regulaçăo da NF-kB pela insulina ainda năo foi elucidada.
voltar |
















;)
;)
;)
;)
;)