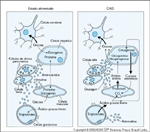
  FIGURA 3-8 FIGURA 3-8
Mecanismos de regulaçăo da glicose. O suprimento de glicose ao cérebro poderá ser mantido por dias e mesmo semanas quando o corpo tiver sido privado de ingestăo calórica. No estado alimentado (esquerda), a assimilaçăo de combustíveis metabólicos e substratos é promovida pela insulina nos tecidos sensíveis ao hormônio. Na cetoacidose diabética (CAD) (direita), hormônios contra-regulatórios (notavelmente o glucagon e a adrenalina) revertem esses processos, promovendo a glicogenólise e criando substratos para a cetogęnese e a neoglicogęnese [14–17]. Negar glicose a tecidos sensíveis ŕ insulina preserva-a para o cérebro. (Adaptado de Kitabchi e Rumbak [17].)
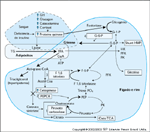 FIGURA 3-9 FIGURA 3-9
Alteraçőes bioquímicas propostas que ocorrem durante a cetoacidose diabética. Estas alteraçőes levam a um aumento de neoglicogęnese e lipólise e uma reduçăo de glicólise. Nota: Ocorre lipólise principalmente no tecido adiposo. Ocorrem outros eventos primariamente no fígado (exceto uma certa neoglicogęnese no rim) [18]. Setas grossas indicam vias estimuladas na cetoacidose diabética, que consistem nas enzimas limitantes de velocidade da neoglicogęnese, enquanto setas finas indicam vias inibitórias na glicólise. O ciclo dos ácidos tricarboxílicos (TCA) é inibido pela inibiçăo da síntese de citrato induzida por acil-graxo e acil coenzima A (CoA), uma enzima limitante de velocidade no ciclo TCA. ATP — trifosfato de adenosina; FFA — free fatty acids [ácidos graxos livres]; F-6-P — frutose-6-fosfato; GH — hormônio de crescimento; G-1-P — glicose-1-fosfato; G-6-P — glicose-6-fosfato; HK — hexoquinase; HMP — hexose monofosfato; PC — piruvato carboxilase; PEP — fosfoenolpiruvato; PEPCK — fosfoenolpiruvato carboxiquinase; PFK — fosfofrutoquinase; PK — piruvato quinase;TG — triglicérides. (Adaptado de Kitabchi et al. [8].)
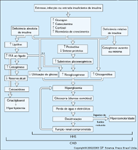 FIGURA 3-10 FIGURA 3-10
Patogęnese da cetoacidose diabética (CAD) e da síndrome hiperglicęmica hiperosmolar (SHH). Alteraçăo do metabolismo de gordura, proteína e carboidrato leva a alteraçőes metabólicas em direçăo aos estados catabólicos e sintomas de poliúria, polidipsia, polifagia, diurese osmótica, desidrataçăo grave e, se năo tratada, coma e óbito. A marca registrada destes eventos é o estado de deficięncia de insulina e aumento de hormônios contraregulatórios. Na SHH, além da relativa deficięncia de insulina e maior desidrataçăo, há também um maior grau de hiperglicemia (secundário a uma menor ingestăo de fluidos) que na CAD. Embora o mecanismo para a ausęncia de um grau significativo de cetose e acidemia na SHH (em comparaçăo com a CAD) năo tenha sido inteiramente elucidado, foi demonstrado em um estudo que o nível do peptídio-C (como uma indicaçăo de reserva de insulina pancreática) é cinco a dez vezes menor na CAD que na SHH [19]. Isto foi oferecido como uma explicaçăo parcial para a ausęncia de cetonemia na SHH. Já que a quantidade necessária de insulina para sua açăo antilipolítica é cerca de cinco a dez vezes menor que para a açăo do transporte de glicose [20], segue-se que a maior a quantidade de insulina residual (peptídio-C) na SHH é suficiente para prevenir lipólise (e, portanto, a ausęncia de cetogęnese na SHH). Esta quantidade de insulina năo é suficiente para promover o transporte de glicose e seus metabolismo; logo, observa-se a hiperglicemia resultante na SHH sem um cetonemia grave. (Adaptado de Kitabchi et al. [4].)
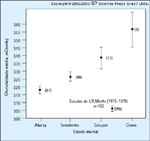 FIGURA 3-11 FIGURA 3-11
Osmolalidade sérica calculada em 122 pacientes com cetoacidose diabética com relaçăo ao estado mental. Cerca de um terço dos pacientes com crises hiperglicęmicas podem se apresentar com um estado mental alterado [6]. Isto pode estar correlacionado com a osmolalidade sérica, mas precisa ser diferenciado de várias afecçőes clínicas associadas a um estado mental alterado ou coma (veja a Fig. 3-7), que podem estar presentes em pacientes diabéticos. (Adaptado de Kitabchi e Fisher [6].)
voltar |
















;)
;)
;)
;)