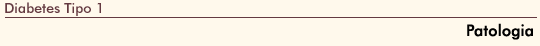  FIGURA 4-11 FIGURA 4-11
(Veja Lâmina Colorida) O efeito do diabetes tipo 1 sobre o pâncreas e células de ilhota. O pâncreas de uma pessoa com diabetes tipo 1 (A) é freqüentemente menor e pesa menos (ie, aproximadamente 50% do peso total do órgăo e 30% do peso endócrino) que seu correspondente sadio (B). Esta diferença é uma conseqüęncia da atrofia progressiva de tecido exócrino que perfaz cerca de 98% do volume pancreático total. C e D demonstram a patologia de amostras pancreáticas de um paciente com diabetes tipo 1 de início recente. Painel C, Ilhota deficiente em insulina corado para glucagon, somatostatina e polipeptídio pancreático. Todas as células endócrinas parecem ter sido coradas, confirmando a ausęncia de células b.
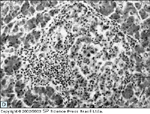 FIGURA 4-11 (Continuação) FIGURA 4-11 (Continuação)
Painel D, Insulite. Um infiltrado de células inflamatórias crônicas está centrado na ilhota. A insulite é uma lesăo difícil de se detectar no pâncreas humano, sendo detectada apenas raras vezes após 1 ano de diabetes tipo 1 patente. Com uma duraçăo prolongada da doença, há o desenvolvimento de uma distorçăo progressiva da arquitetura da ilhota, com uma tendęncia de células a e d saírem da ilhota e se espalharem como células isoladas no paręnquima exócrino. (Painel C, imunocoloraçăo com fosfatase alcalina, X1.150; painel D, coloraçăo com hematoxilina-eosina, X300.) (Painéis C e D de Foulis [8]; com permissăo.)
 FIGURA 4-12 FIGURA 4-12
(Veja Lâmina Colorida) Ilhota de um paciente com diagnóstico recente de diabetes tipo 1 demonstrando uma infiltraçăo linfocítica difusa (insulite) com início de atrofia nos cordőes de ilhota. (Coloraçăo com hematoxilina-eosina, X300.) (De Foulis [8]; com permissăo.)
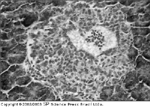 FIGURA 4-13 FIGURA 4-13
(Veja Lâmina Colorida) Patologia de uma ilhota em regeneraçăo no pâncreas de um paciente com diagnóstico recente de diabetes tipo 1. Células de ilhota recém-formadas derivam do epitélio de um ducto. Linfócitos estăo presentes na luz do ducto e em alguns lugares da periferia. Evidęncias de tal regeneraçăo săo raras nos órgăos pancreáticos de pacientes com diabetes tipo 1 e estăo geralmente limitadas ŕqueles que morrem logo depois do início da doença. (Coloraçăo com hematoxilina-eosina, X400.) (De Foulis [8]; com permissăo.)
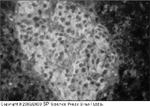 FIGURA 4-14 FIGURA 4-14
(Veja Lâmina Colorida) Auto-anticorpos anti-célula de ilhota (ICA – islet cell autoantibodies). Auto-anticorpos anti-célula de ilhota estăo presentes no soro de aproximadamente 75% das pessoas no início do diabetes tipo 1 versus 0,4% de pessoas saudáveis. Primeiro auto-anticorpo atribuído ao diabetes tipo 1, a presença de auto-anticorpos anti-célula de ilhota é identificada por ensaio imuno-fluorescente indireto que utiliza pâncreas do grupo sangüíneo O humano. Os auto-anticorpos anti-célula de ilhota específicos para células b foram identificados. Entretanto, os auto-anticorpos reagem com todas as células dentro da ilhota, inclusive aquelas que secretam insulina (células b), glucagon (células a), células de somatostatina (células d) e polipeptídio pancreático (células PP). Os auto-antígenos até agora considerados responsáveis pela reaçăo de ICA incluem sialoglicolípides, ácido glutâmico descarboxilase e ICA512/IA-2.
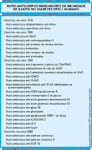 FIGURA 4-15 FIGURA 4-15
Auto-anticorpos marcadores de imunidade de ilhota no diabetes tipo 1 humano. Desde a primeira descriçăo dos auto-anticorpos anti-célula de ilhota (ICAs – islet cell autoantibodies) em 1974 (veja Fig. 4-14), muitos novos auto-anticorpos marcadores de imunidade anti-ilhota foram identificados em pacientes com diabetes tipo 1. Além de sua presença no início da doença, muitos desses marcadores se relevaram úteis na identificaçăo de pacientes no período pré-sintomático, meses a anos antes do início clínico do diabetes tipo 1. Destes, quatro marcadores conquistaram grande aceitaçăo por causa da confirmaçăo científica, alta freqüęncia de expressăo e sensibilidade e especificidades superiores da doença: ICA, auto-anticorpos antiinsulina, ácido glutâmico descarboxilase e auto-anticorpos anti-IA-2.
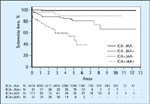 FIGURA 4-16 FIGURA 4-16
Uso de auto-anticorpos contra auto-antígenos de célula de ilhota para predizer casos futuros de diabetes tipo 1. Esta análise de tabela da vida indica a probabilidade de permanecer livre da doença, estratificada pelo aparecimento de auto-anticorpos citoplasmáticos anti-célula de ilhota (ICA – islet cell cytoplasmic autoantibodies) e auto-anticorpos antiinsulina (IAA – insulin autoantibodies) em parentes de probandos (casos índice) com a doença. Na parte inferior, é mostrado o número de parentes acompanhados desde a identificaçăo de auto-anticorpos, para cada grupo. Como se pode observar, a probabilidade de desenvolvimento de diabetes tipo 1 é máxima naquelas pessoas com dois auto-anticorpos, com aproximadamente metade dessas pessoas desenvolvendo a doença num prazo de 4 anos. (Adaptado de Krischer et al. [9].)
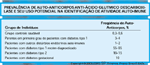 FIGURA 4-17 FIGURA 4-17
Prevalęncia de auto-anticorpos anti-ácido glutâmico descarboxilase e seu uso potencial na identificaçăo de atividade auto-imune. Os anticorpos anti-ácido glutâmico descarboxilase (GAD – glutamic acid decarboxylase) servem de marcador para prediçăo de casos futuros e para diagnóstico de novos casos de diabetes tipo 1. Investigaçőes recentes aqui resumidas de forma representativa indicam que os auto-anticorpos anti-GAD também podem ser úteis na identificaçăo de auto-imunidade em pessoas com diagnóstico de outras formas de diabetes. Estas identificaçőes podem ser úteis em termos de oferecer um tratamento e assistęncia clínica apropriados do diabetes.
 FIGURA 4-18 FIGURA 4-18
Modelo de camundongo diabético năo-obeso de diabetes tipo 1.
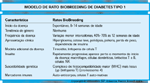 FIGURA 4-19 FIGURA 4-19
Modelo de rato BioBreeding de diabetes tipo 1.
 FIGURA 4-20 FIGURA 4-20
(Veja Lâmina Colorida) Estágios de desenvolvimento da lesăo de insulite em camundongos diabéticos năo-obesos. Patologia de amostras pancreáticas de célula normal de ilhota sem infiltrado leucocítico (A) e em vários estágios de infiltraçăo (B a H).
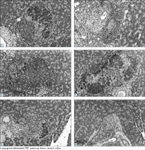 FIGURA 4-20 (Continuação) FIGURA 4-20 (Continuação)
A partir de 5 a 7 semanas de idades, os leucócitos circundam e acabam infiltrando as ilhotas em números crescentes. A partir de 12 a 14 semanas, esta insulite inicial (freqüentemente denominada năo-destrutiva) é substituída por um insulite que destrói as células b produtoras de insulina. Quando a ilhota está destituída de células b, desaparece o infiltrado leucocítico, deixando apenas células a, t e d (painel H). (Coloraçăo com hematoxilina-eosina seguida de contra-coloraçăo com anticorpo antiinsulina e avidina- biotina, X300.) (Cortesia de A. Peck, University of Florida.)
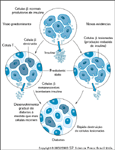 FIGURA 4-21 FIGURA 4-21
Um novo modelo mostrando uma rápida destruiçăo de células b na patogęnese do diabetes tipo 1. Até recentemente, a maioria dos modelos que avaliam a velocidade de destruiçăo de células b presumiu uma perda gradual (ie, linear modificado; veja Fig. 4-2) de células endócrinas, caracterizada por pequenos períodos de “aumento e diminuiçăo” da resposta imune. Entretanto, investigaçőes recentes de camundongos diabéticos năo-obesos sugeriram que a destruiçăo real de células b ocorre em um período de tempo bem limitado imediatamente antes do início sintomático. A composiçăo da lesăo insulítica, da atividade destrutiva ou de ambas antes deste evento seria de capacidade năo-destrutiva ou de capacidade destrutiva limitada. Embora existam evidęncias limitadas para este modelo em termos do diabetes tipo 1 humano, elas săo tema de investigaçăo em andamento.
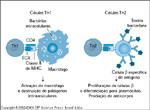 FIGURA 4-22 FIGURA 4-22
Modelo Th1/Th2 de imunorregulaçăo. Estudos in vivo e in vitro corroboram a noçăo de que atividades de células “helper-T” CD4+ possam estar direta ou indiretamente relacionadas ŕ produçăo de citocinas específicas. Imunologistas evolucionários sugerem que a compartimentalizaçăo de tais respostas propicia um desenvolvimento mais eficiente de uma resposta imune contra patógenos de origens e modos de evasăo divergentes. Embora uma generalizaçăo um tanto exagerada, considera-se que as citocinas Th1 amplificariam as atividades imunológicas celulares, enquanto as citocinas Th2 apoiariam as da imunidade humoral. Especificamente, a atividade de Th1 parece ser intensificada pela produçăo das linfocinas interferon g (IFN-g), interleucina 2 (IL-2) e IL-12. Reciprocamente, ocorre reforço pela Th2 da imunidade humoral através da liberaçăo de IL-4 e IL-10. Note que a IL-4 parece ser um forte inibidor da imunidade Th1. A produçăo de citocinas específicas, tanto sistęmicas como no local da inflamaçăo pancreática, pode ter importantes implicaçőes para a patogęnese do diabetes bem como para o potencial de desenvolvimento de métodos voltados para a prevençăo da doença (veja Fig. 4-30). MHC — complexo de histocompatibilidade maior; TCR — T-cell receptor [receptor de célula T].
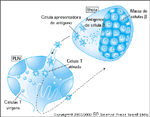 FIGURA 4-23 FIGURA 4-23
Modelo hipotético para a iniciaçăo do diabetes tipo 1. Células T virgens circulam através do sangue e órgăos linfóides, inclusive nódulos linfáticos pancreáticos (NLP). Nos nódulos, elas encontram as células apresentadoras de antígeno (bem provavelmente células dendríticas) que exibem em sua superfície moléculas do complexo de histocompatibilidade maior (MHC) portando antígenos na forma de fragmentos peptídicos. Neste caso, os antígenos derivam de proteínas sintetizadas pelas células b de ilhota pancreática, apanhadas (na forma solúvel, como pedaços celulares ou como células apoptóticas) quando as células apresentadoras de antígenos residiam nas ilhotas. Uma fraçăo diminuta das células T virgens reconhece complexos de molécula de MHC/antígeno de célula b, torna-se ativada e, entăo, acessa os tecidos, inclusive o pâncreas, onde se reencontra com o antígeno cognato, é reativada e é retida. (Adaptado de Mathis et al. [10].)
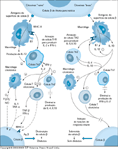 FIGURA 4-24 FIGURA 4-24
O modelo da citocina “boa e ruim” para a patogęnese do diabetes tipo 1. Segundo o modelo criado de acordo com as Figuras 4-22 e 4-23, considera-se que a produçăo de citocinas Th2 fornece uma via de esquiva da destruiçăo de células b (e, portanto, o rótulo de citocina “boa”). A produçăo de interleucina 4 (IL-4) e IL-10 bloquearia as açőes destrutivas da resposta imune celular. Em contraste, seria considerado que as respostas imunes caracterizadas pelas citocinas Th1 “ruim” promovem a destruiçăo de células b através da intensificaçăo de açőes imputadas ŕs células T citotóxicas ou aos macrófagos (através de lesăo mediada por radical de oxigęnio). Ag — antígeno; H2 O2 — peróxido de hidrogęnio; IFN — interferon; MHC — complexo de histocompatibilidade maior; sinal de menos — via inibitória; NO — óxido nítrico; O2 — radical livre de oxigęnio; sinal de mais — via promotora;TNF-a — fator-a de necrose tumoral.
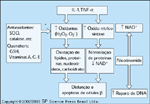 FIGURA 4-25 FIGURA 4-25
O modelo de linfocinas para destruiçăo de célula b no diabetes tipo 1. Neste cenário, as linfocinas produzidas em resposta a uma infecçăo inespecífica iriam conferir, por seu modo de açăo, um grau limitado de destruiçăo inicial de células b devido a uma suscetibilidade fora do comum das células b a tais agentes. As linfocinas predominantes citadas neste modelo săo aquelas produzidas por macrófagos e incluem o fator-a de necrose tumoral (TNF-a) e a interleucina 1 (IL-1). Apesar dos benefícios da recuperaçăo da infecçăo, este processo de imunidade anti–célula b resultaria naqueles geneticamente suscetíveis ŕ doença. O processo contínuo de destruiçăo de células b ocorreria através da açăo de vários agentes citotóxicos (eg, oxidantes e óxido nítrico) bem como de uma falta de atividade de inúmeros compostos associados a mecanismos de reparo celular ou de DNA (eg, anti-oxidantes). GSH — hormônio glomeruloestimulante; H2 O2 — peróxido de hidrogęnio; NAD+ — nicotina adenina dinucleotídeo oxidado; O2 - — radical livre de oxigęnio; SOD — superóxido dismutase. (Adaptado de Kolb e Kolb-Bachofen [11].)
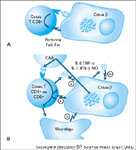 FIGURA 4-26 FIGURA 4-26
Mecanismos propostos de morte de célula b. A, Num modelo baseado em reconhecimento, uma célula T CD8+ é ativada por reconhecimento direto do antígeno de célula b de ilhota (círculos) apresentado pelas moléculas do complexo de histocompatibilidade maior nas células b. A ativaçăo provoca a morte da célula b através de contato célula/célula que utiliza várias vias (eg, Fas/FasL, perforina). B, Num modelo baseado em ativaçăo, uma célula T (CD4+ ou CD8+ ) reconhece antígenos de célula b apresentados indiretamente pelas células apresentadoras de antígeno (CAA) localizadas nas vizinhanças da ilhota. A ativaçăo resultante provoca morte da célula b por: (i) receptores de superfície (eg, Fas/FasL), (ii) mediadores solúveis provenientes de células T, (iii) ativaçăo de atividades citocidas do macrófago, ou (iv) ativaçăo de sinais de morte nas células b. IFN — interferon; IL — interleucina; NO — óxido nítrico. (Adaptado de Mathis et al. [10].)
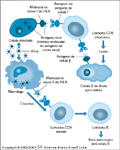 FIGURA 4-27 FIGURA 4-27
Papel potencial para vírus na patogęnese do diabetes tipo 1: o modelo do mimetismo molecular. Neste modelo, o processo auto-imune começa depois de uma resposta imune “normal” a uma célula infectada por um vírus cujas proteínas compartilham uma seqüęncia similar ŕquela de uma proteína de célula b. As células infectadas exibem os antígenos virais processados (por meio de moléculas da Classe I) ŕs células T CD8+. Os macrófagos, tendo fagocitado e processado os vírus, apresentam os peptídios virais ŕs células T CD4+ através de moléculas da Classe II. As células T CD4+ amplificam as açőes das células T CD8+ para se tornarem células efetoras citotóxicas que podem matar células b que expressam um peptídio comum ŕ proteína viral. Apesar de esforços exaustivos de pesquisa para demonstrar o mimetismo molecular como uma causa de base do diabetes tipo 1, ele continua um modelo năo-comprovado. A corroboraçăo contemporânea deriva predominantemente de estudos que demonstram similaridade em seqüęncia de aminoácidos entre proteínas de células b (eg, ácido glutâmico descarboxilase e IA-2) e aquelas de vírus (eg, Coxsackie e Rotavírus) e a capacidade de moléculas do antígeno linfocitário humano (HLA) com suscetibilidade e resistęncia ŕ doença do tipo I de se ligar a essas regiőes de mimetismo. Estudos, em particular os de imunidade celular, da história natural do diabetes em seres humanos e camundongos diabéticos năo-obesos năo conseguiram elevar este modelo além do estágio hipotético. (Adaptado de Atkinson e Eisenbarth [1].)
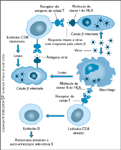 FIGURA 4-28 FIGURA 4-28
Papel potencial para vírus na patogęnese do diabetes tipo 1: modelos de vírus b-trópico–superantígeno viral. Nestes modelos, a iniciaçăo do processo auto-imune ocorre depois de uma infecçăo viral direta de células b ou da expressăo em células b de um vírus que age como um superantígeno. Nas duas situaçőes, os leucócitos săo recrutados para as ilhotas pancreáticas. O recrutamento aumenta a liberaçăo de citocinas (eg, interferon-a) e a adesăo de leucócitos dentro das ilhotas pancreáticas. No modelo de tropismo pela célula b, a célula b infectada é suscetível a um ataque direto pelos linfócitos citotóxicos antivirais. Nos dois modelos, as citocinas e os radicais livres produzidos por macrófagos ativados dentro das células de ilhota podem aumentar a resposta citotóxica ŕs células b; as citocinas também recrutam células T CD4+ para a lesăo. Os macrófagos apresentam auto-antígenos derivados de células b danificadas pelos vírus, levando desta forma ao desenvolvimento de linfócitos e auto-anticorpos que reagem com proteínas de célula b. Existe corroboraçăo para estes dois modelos, embora eles continuem hipotéticos. Embora vírus capazes de destruir células b tenham sido isolados de um número extremamente limitado de órgăos pancreáticos humanos, o exame de um grande número desses tecidos de pacientes com diabetes tipo 1 năo foi capaz de revelar a presença de tais vírus. Existe corroboraçăo para o modelo do superantígeno através da identificaçăo de células T características da ativaçăo do superantígeno nos órgăos pancreáticos de um número limitado de pacientes com diabetes tipo 1 de início recente. Até agora, contudo, nenhum superantígeno viral desse tipo foi identificado de maneira indiscutível. HLA — antígeno leucocitário humano. (Adaptado de Atkinson e Eisenbarth [1].)
voltar |
















;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)