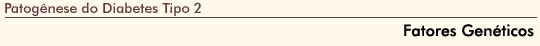  FIGURA 6-8 FIGURA 6-8
Concentraçőes plasmáticas de glicose (topo) e insulina (embaixo) em experimentos de clamp hiperglicęmico. Em resposta a uma elevaçăo aguda de concentraçőes sangüíneas de glicose, a insulina é secretada de maneira bifásica, uma primeira fase durando aproximadamente 10 minutos seguida por uma segunda fase gradualmente crescente. A primeira fase da liberaçăo de insulina foi vinculada a grânulos de insulina localizados perto da membrana de célula b (pool rapidamente liberável). A liberaçăo da insulina da segunda fase depende em parte da mobilizaçăo dos grânulos de insulina de um pool de armazenamento para um pool rapidamente liberável, bem como de síntese aumentada de insulina. Neste estudo [15], indivíduos com tolerância normal ŕ glicose mas com um parente em primeiro grau com diabetes tipo 2 foram estudados com o uso de um clamp hiperglicęmico para avaliar sua funçăo de célula b e sensibilidade ŕ insulina em relaçăo a um grupo de indivíduos com tolerância normal ŕ glicose, mas sem história familiar de diabetes. Os indivíduos eram equivalentes quanto ŕ idade, gęnero e obesidade para excluir fatores de risco ambientais (adquiridos). Foi demonstrado que indivíduos com um parente em primeiro grau com diabetes tipo 2 apresentaram reduçăo na liberaçăo precoce (primeira fase) e tardia (segunda fase) da insulina e năo eram resistentes ŕ insulina. (Adaptado de Pimenta et al. [15].)
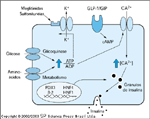 FIGURA 6-9 FIGURA 6-9
Detecçăo de nutrientes e secreçăo de insulina pela célula b pancreática. A célula b capta glicose e aminoiácidos via transportadores específicos na membrana celular, como o transportador de glicose GLUT2. Esta isoforma do transportador só é expressa pela célula b e pelo fígado e tem uma Km na faixa fisiológica. Uma vez dentro da célula, a glicose é fosforilada por uma forma especializada de hexoquinase chamada glicoquinase. O metabolismo subseqüente da glicose resulta em uma alteraçăo na razăo trifosfato de adenosina (ATP):difosfato de adenosina (ADP) dentro da célula que, por sua vez, causa ativaçăo do canal de potássio sensível ao ATP. Isto resulta na despolarizaçăo da célula, em um influxo de cálcio e a subseqüente liberaçăo da insulina dos grânulos secretores. O receptor de sulfoniluréia também pode ativar o canal de potássio sensível ao ATP, mimetizando o efeito da glicose. Outros secretagogos, como o peptídio-1 semelhante ao glucagon (GLP-1), se desviam deste sistema através da alteraçăo dos níveis celulares de monofosfato de adenosina cíclico (cAMP). O nível de expressăo das várias moléculas envolvidas na detecçăo de glicose, inclusive o transportador de glicose GLUT2, e o desenvolvimento da célula b săo controlados por vários fatores de transcriçăo nucleares. Destes, os mais bem estudados săo o HNF-1a, o HNF-1b, o HNF-4b e o PDX-1 (IPF-1). O diabetes da juventude com início na maturidade pode resultar de defeitos genéticos em qualquer um destes fatores de transcriçăo ou um defeito genético na glicoquinase. No diabetes tipo 2, o local exato do defeito na detecçăo de glicose é desconhecido, mas estudos em modelos animais da doença sugeriram que isto pode ser o resultado de uma regulaçăo para baixo ['down-regulation'] do transportador de glicose GLUT2 [75].
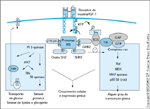 FIGURA 6-10 FIGURA 6-10
A rede de sinalizaçăo da insulina. A rede completa é complexa e pode ser dividida em cinco níveis: 1) ativaçăo da tirosina quinase do receptor de insulina e eventos intimamente ligados; 2) fosforilaçăo de uma família de proteínas de substrato; 3) interaçăo do receptor e seus substratos com várias moléculas de sinalizaçăo intermediárias via SH2 (src homology 2) e outros domínios de reconhecimento; 4) ativaçăo da serina e lípide quinases, resultando numa ampla gama de eventos de fosforilaçăo-desfosforilaçăo; e 5) regulaçăo dos efetores biológicos finais da açăo da insulina, como transporte de glicose, síntese lipídica, expressăo gęnica e mitogęnese. As proteínas SH2 ligam as proteínas de substrato do receptor de insulina (IRS – insulin receptor substrate) a uma série de reaçőes em cascata envolvendo serina/treonina quinases e fosfatases, como as proteínas quinases ativadas por mitógeno (MAP kinases), S6 quinases e proteína fosfatase-1A. Estas serina quinases agem sobre enzimas como a glicogęnio sintase, fatores de transcriçăo e outras proteínas para produzir muitos dos efeitos biológicos finais do hormônio. No tecido adiposo e músculo, a estimulaçăo pela insulina também aumenta a captaçăo de glicose, promovendo a translocaçăo de um pool intracelular de transportadores de glicose até a membrana plasmática. Năo se sabe como exatamente esta açăo é ligada ŕ cascata de fosforilaçăo, mas vários estudos sugerem que esta importante açăo da insulina, bem como a maioria dos efeitos metabólicos, está a jusante da enzima fosfatidilinositol 3-quinase (PI 3-quinase). Outros efeitos da insulina, como a estimulaçăo de síntese de glicogęnio e lípides, ocorrem através de efeitos intracelulares adicionais para estimular as enzimas envolvidas nestas reaçőes. ATP — trifosfato de adenosina; GAP — proteína ativadora de GTPase; GRB2 -proteína-2 de ligaçăo ao receptor do fator de crescimento; GTP – trifosfato de guanosina; IGF — fator de crescimento semelhante ŕ insulina; MEK — MAP-Erk quinase; SOS — proteína son-of-sevenless.
 FIGURA 6-11 FIGURA 6-11
Resistęncia ŕ insulina e obesidade. A resistęncia ŕ insulina associada ao diabetes tipo 2 pode ser basicamente explicada pela obesidade, já que indivíduos comparavelmente obesos com e sem diabetes tipo 2 tęm reduçőes quantitativamente semelhantes na ligaçăo ao receptor de insulina, na atividade da tirosina quinase do receptor de insulina e no transporte muscular de glicose [42,76]. A maioria destas anormalidades em pacientes com diabetes tipo 2 pode ser normalizada por perda de peso [77]. Evidęncias atuais indicam que o fator mais importante envolvido na resistęncia ŕ insulina da obesidade em seres humanos săo os níveis circulantes aumentados de ácidos graxos livres (FFA – free fatty acids) plasmáticos [43]. Uma elevaçăo experimental de FFA em seres humanos normais diminui a captaçăo muscular de glicose e aumenta a produçăo endógena de glicose e, em modelos animais, compromete a secreçăo de insulina [78,79]. Todas estas açőes promoveriam a hiperglicemia através da alteraçăo das taxas de equilíbrio entre entrada e remoçăo da glicose da circulaçăo. Săo dois os mecanismos para os efeitos do FFA: competiçăo pelo substrato [80] e comprometimento da sinalizaçăo da insulina [81]. Produtos acetilados do metabolismo de FFA reduzem a oxidaçăo da glicose via inibiçăo da piruvato desidrogenase e ativam as serina/treonina quinases, possivelmente através de aumentos na proteína quinase C (theta), levando a uma atividade reduzida das proteínas do substrato do receptor de insulina (IRS – insulin receptor substrate). O principal efeito destas últimas parece ser uma reduçăo no transporte de glicose [82-84].
 FIGURA 6-12 FIGURA 6-12
Estágios no desenvolvimento do diabetes tipo 2. Estudos longitudinais e transversais indicam que os indivíduos destinados a desenvolver diabetes tipo 2 passam por cinco estágios. O primeiro estágio começa ao nascimento, quando a homeostase da glicose é normal, mas os indivíduos encontram-se sob risco de diabetes tipo 2 por causa dos polimorfismos genéticos que os predispőem a se tornarem obesos e que limitam a capacidade das células b pancreáticas de compensar a resistęncia ŕ insulina. Durante o estágio 2, reduçőes na sensibilidade insulina emergem como resultado de uma predisposiçăo genética e de estilo de vida năo-saudável, que săo inicialmente compensadas por um aumento na funçăo de células b de maneira que a tolerância ŕ glicose permanece normal. Durante o estágio 3, a funçăo de célula b e a sensibilidade ŕ insulina deterioram, de tal maneira que, quando desafiadas, como durante um teste de tolerância ŕ glicose ou uma refeiçăo padronizada, a tolerância ŕ glicose pós-prandial se torna anormal. Neste ponto, a funçăo da célula b é claramente anormal, mas suficiente para manter concentraçőes normais de glicose plasmática em jejum. No estágio 4, como resultado de deterioraçăo adicional no funcionamento de células b e agravamento da sensibilidade ŕ insulina (provavelmente como resultado da hiperglicemia pós-prandial), as concentraçőes plasmáticas de glicose em jejum aumentam por causa de um aumento na produçăo basal endógena de glicose. Finalmente, no estágio 5, como resultado de uma deterioraçăo adicional na funçăo de células b (por causa de fatores genéticos e ambientais, como glico e lipotoxicidade), os níveis de glicose tanto em jejum como pós-prandiais atingem níveis diabéticos. IFG – impaired fasting glucose [comprometimento da glicose em jejum];TGD – tolerância ŕ glicose diminuída.
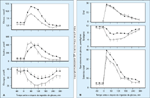 FIGURA 6-13 FIGURA 6-13
Comparaçăo das alteraçőes em hormônios (insulina e glucagon) e taxas de produçăo e utilizaçăo de glicose após ingestăo de glicose em pacientes com tolerância ŕ glicose diminuída (círculos fechados) e em pacientes saudáveis (círculos abertos). Pacientes com tolerância ŕ glicose diminuída tęm níveis plasmáticos normais em jejum de glicose, insulina e glucagon e taxas normais em jejum de produçăo e utilizaçăo de glicose. Entretanto, após um desafio de glicose oral ou uma refeiçăo, eles apresentam uma liberaçăo inicial reduzida de insulina durante os primeiros 30 a 60 minutos, acompanhada por um menor decréscimo nos níveis plasmáticos de glucagon (A). Estas anormalidades hormonais levam a uma reduçăo na supressăo da produçăo endógena de glicose com preservaçăo do seqüestro esplâncnico normal da glicose ingerida. Conseqüentemente, uma quantidade de glicose acima do normal entra na circulaçăo sistęmica (B). Isto excede as taxas de remoçăo de glicose da circulaçăo durante 1 a 2 horas iniciais, de forma que os níveis plasmáticos de glicose aumentam mais que o normal. A hiperglicemia finalmente leva a níveis plasmáticos tardios e maiores que o normal de insulina que, juntamente com a hiperglicemia, fazem com que a utilizaçăo de glicose exceda a produçăo de glicose e, portanto, fazem com que a glicose plasmática acabe voltando aos níveis normais em jejum [85,86]. Durante o período pós-prandial de 4 a 6 horas, uma quantidade maior que o normal de glicose entrou na circulaçăo e os níveis plasmáticos de glicose começaram em um nível normal e voltaram a um nível normal; portanto, é óbvio que uma quantidade de glicose maior que o normal foi removida da circulaçăo. Embora a utilizaçăo tissular de glicose năo esteja reduzida em pacientes com tolerância ŕ glicose diminuída (TGD), ela é menor que aquela que teria sido encontrada em pacientes com tolerância normal ŕ glicose cujos níveis plasmáticos de glicose e insulina eram equivalentes ŕqueles de pacientes com TGD, indicando que pacientes com TGD săo resistentes ŕ insulina [87]. (Adaptado de Mitrakou et al. [86].)
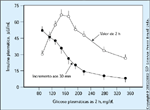 FIGURA 6-14 FIGURA 6-14
Comparaçăo de respostas precoces e tardias de insulina plasmática durante testes orais de tolerância ŕ glicose. A reduçăo na secreçăo pós-prandial precoce (30 min) de insulina apresentou correlaçăo com a supressăo reduzida da produçăo endógena de glicose [86] e diminui progressivamente ŕ medida que deteriora a tolerância ŕ glicose [87]. Em contraste, os níveis de insulina após 2 horas aumentam inicialmente e só diminuem depois que os níveis plasmáticos de glicose após 2 horas atingem valores diabéticos [88]. Foi erroneamente interpretado que este último fenômeno implicava que a resistęncia ŕ insulina ocorreria antes do comprometimento da secreçăo de insulina na evoluçăo do diabetes tipo 2 [89], sendo agora evidente que isto resulta da hiperglicemia decorrente de uma liberaçăo inicial atrasada da insulina [6,7,88,90].
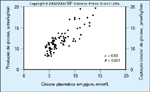 FIGURA 6-15 FIGURA 6-15
Produçăo e captaçăo da glicose no estado pós-absortivo. Com uma deterioraçăo adicional da funçăo de células b, os níveis plasmáticos de glicose no estado de jejum aumentam como resultado da supressăo comprometida da liberaçăo hepática e renal de glicose pela insulina [52]. Isto se deve basicamente a um aumento na neoglicogęnese [91-94]. Como mostra esta figura, a remoçăo de glicose da circulaçăo também aumenta ŕ medida que aumentam os níveis sangüíneos de glicose em jejum, ilustrando que a causa primária da hiperglicemia de jejum é a superproduçăo de glicose, e năo uma reduçăo na utilizaçăo de glicose. A produçăo aumentada de glicose resulta do comprometimento da funçăo de células b, bem como da resistęncia ŕ insulina, mediados em parte indiretamente pelos níveis plasmáticos aumentados de ácidos graxos, pela disponibilidade aumentada de substratos gliconeogęnicos e pela ausęncia de uma supressăo adequada da secreçăo de glucagon. (Adaptado de Dinneen et al. [52].)
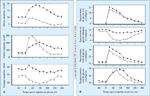 FIGURA 6-16 FIGURA 6-16
Comparaçăo de alteraçőes pós-prandiais nos níveis plasmáticos de insulina e glucagon (A) e taxas de aparecimento e remoçăo de glicose do plasma (B) em pacientes com diabetes tipo 2 e pacientes saudáveis. As anormalidades pós-prandiais em pacientes com diabetes tipo 2 (círculos fechados) săo bem semelhantes ŕquelas de pessoas com tolerância ŕ glicose diminuída (círculos abertos), exceto que săo mais exageradas. A única diferença importante é que, com glicosúria excessiva, a captaçăo de glicose no fígado está diminuída [51,52,95,96]. (Adaptado de Mitrakou et al. [51].)
voltar |
















;)
;)
;)
;)
;)
;)
;)
;)
;)